Poter prevedere, visualizzare e creare nuove proteine ha portato ad un'accelerazione nello sviluppo di nuovi farmaci. Un successo possibile grazie a software e modelli di AI messi a punto dai Nobel per la chimica 2024
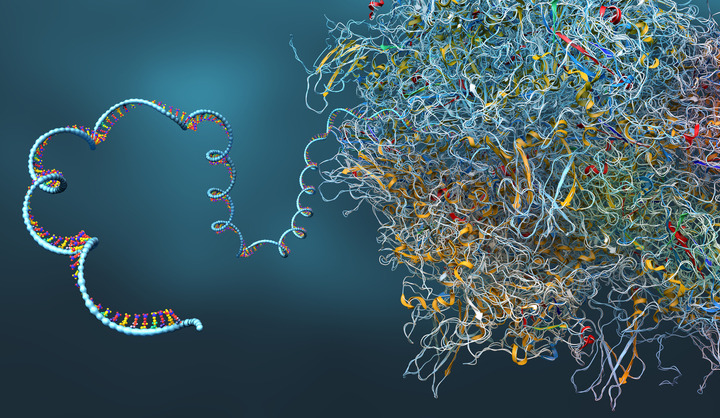
A David Baker, Demis Hassabis e John M. Jumper il Nobel per la chimica 2024. A loro il merito di aver messo a punto software e modelli di intelligenza artificiale in grado di prevedere la struttura delle proteine e realizzarne di nuove, premessa fondamentale per la comprensione di ciò che avviene nel nostro organismo e per sviluppare nuovi farmaci. Un premio, dunque, che sa molto di medicina.
A COSA SERVE STUDIARE LE PROTEINE?
Le proteine sono macromolecole formate da catene di amminoacidi legate in sequenza e ripiegate in enormi strutture tridimensionali complesse. Ogni proteina svolge specifiche funzioni biologiche come, ad esempio, regolare i processi cellulari, catalizzare reazioni chimiche, trasportare molecole e fornire struttura ai tessuti. Comprenderne la forma è di fondamentale importanza. Molte di esse infatti agiscono grazie ad un meccanismo simile ad una chiave nella sua serratura. In passato lo studio sulla conformazione delle proteine era un processo lungo e complesso basato principalmente su tecniche come la cristallografia a raggi X e la risonanza magnetica nucleare. Questi metodi richiedevano un particolare pre-trattamento e la successiva analisi tramite una sorta di "radiografia". Un processo che poteva richiedere anni per ottenere risultati su una singola proteina. Non solo, con le tecniche del passato si poteva ottenere solo un'istantanea e non in che modo la proteina in questione può cambiare la sua forma interagendo con altre strutture.
PREVEDERE LA STRUTTURA TRIDIMENSIONALE NEL DETTAGLIO
A cambiare radicalmente l'approccio allo studio della struttura delle proteine ci ha pensato la progressiva potenza di calcolo dei computer. Ma la potenza non è tutto. Occorreva mettere a punto un sistema di calcolo. Demis Hassabis e John Jumper, con il loro team di Google DeepMind, hanno sviluppato AlphaFold, un modello di intelligenza artificiale capace di prevedere con precisione la struttura tridimensionale delle proteine a partire dalla loro sequenza di amminoacidi. Negli anni sono state sviluppate diverse versioni come AlphaFold2 e 3 da poco rilasciato. Grazie ad essi le oltre 200 milioni di proteine conosciute possono essere visualizzate con un'accuratezza paragonabile ai metodi sperimentali classici. David Baker invece, realizzando il software Rosetta, ha sviluppato metodi per creare proteine che non esistono in natura partendo dalla definizione di una struttura desiderata e progettando una sequenza di amminoacidi che possa piegarsi in quella forma.
LE RICADUTE IN CAMPO MEDICO
Poter prevedere e visualizzare un'infinita quantità di proteine ha portato ad un'improvvisa accelerazione della ricerca in campo biomedico e non solo. Uno dei principali è la progettazione di nuove terapie: la conoscenza dettagliata della struttura delle proteine consente di progettare farmaci che si adattano perfettamente alle strutture su cui devono agire. E non è solo questione di precisione ma anche di tempo. Grazie ad AlphaFold2 è possibile ottenere informazioni in pochi giorni velocizzando lo sviluppo preclinico dei nuovi farmaci. La possibilità di prevedere in anticipo la conformazione di una proteina aiuta infatti a limitare i costosi esperimenti necessari a verificare il meccanismo di azione di una determinata terapia.
PROGETTARE NUOVE PROTEINE
Ma poterle anche progettare, oltre che prevedere, è stato un salto quantico nella storia della medicina. Un esempio pratico è la creazione di anticorpi monoclonali (che sono proteine) utili a trattare numerose patologie come il cancro. Gli immunoterapici in commercio sono tutti anticorpi monoclonali, per intenderci. Anche se la capacità di crearne di nuovi viene prima dello sviluppo di Rosetta, il software sviluppato dal Nobel Baker ha il merito di poter essere utilizzato per studiare e ottimizzare la "forma" di alcune zone particolari delle proteine. In altre parole, con questo approccio è possibile migliorare l'affinità tra l'anticorpo e il suo bersaglio. Un dettaglio non indifferente nello sviluppo, ad esempio, delle Car-T dove è fondamentale che il recettore inserito nei linfociti T riconosca la proteina tumorale posta sulle cellule cancerose. Rosetta dunque, secondo gli addetti ai lavori, è quel software in grado di offrire un approccio computazionale utile sia a migliorare le caratteristiche delle molecole già esistenti o per crearne di nuove con funzioni personalizzate. Un approccio rivoluzionario dal duplice effetto: realizzare farmaci più efficaci in minor tempo.
Sostieni la ricerca, sostieni la vita. Dona ora.
Dona ora per la ricerca contro i tumori

Fonti

Daniele Banfi
Giornalista professionista del Magazine di Fondazione Umberto Veronesi dal 2011. Laureato in Biologia presso l'Università Bicocca di Milano - con specializzazione in Genetica conseguita presso l'Università Diderot di Parigi - ha un master in Comunicazione della Scienza ottenuto presso l'Università La Sapienza di Roma. In questi anni ha seguito i principali congressi mondiali di medicina (ASCO, ESMO, EASL, AASLD, CROI, ESC, ADA, EASD, EHA). Tra le tante tematiche approfondite ha raccontato l’avvento dell’immunoterapia quale nuova modalità per la cura del cancro, la nascita dei nuovi antivirali contro il virus dell’epatite C, la rivoluzione dei trattamenti per l’ictus tramite la chirurgia endovascolare e la nascita delle nuove terapie a lunga durata d’azione per HIV. Dal 2020 ha inoltre contribuito al racconto della pandemia Covid-19 approfondendo in particolare l'iter che ha portato allo sviluppo dei vaccini a mRNA. Collabora con diverse testate nazionali.